
Abstract
Remedial systems for Alzheimer’s illness (AD) still can’t seem to offer an illness-altering impact to stop the crippling movement of neurodegeneration and mental deterioration. Rather, medicines so far are restricted to specialists that sluggish infection movement without stopping it, and albeit much work towards a fix is in progress, a more prominent comprehension of illness etiology is positively important for any such accomplishment. Mitochondria, as the focus of cell metabolic action and the essential generators of receptive oxidative species in the cell, got specific consideration particularly given that mitochondrial deserts are known to add to cell harm. Besides, as oxidative pressure has come to the front of AD as a causal hypothesis, and as mitochondrial harm is known to go before a large part of the trademark pathologies of AD, it appears to be progressively obvious that this metabolic organelle is at last answerable for much, while possibly not all of the illness pathogenesis. In this audit, we survey the job of neuronal mitochondria in the pathogenesis of AD and fundamentally evaluate treatment procedures that use this upstream passage as a strategy for sickness avoidance. We presume that with a resuscitated spotlight on mitochondrial fix and insurance, a powerful and practical helpful specialist can be effectively evolved.
Introduction
Mitochondrial anomalies have for quite some time been ensnared in the maturing system, yet as of late has their impact been reached out to neurodegenerative sickness [1,2,3,4]. Alzheimer’s infection (AD), specifically, seems to include the inevitable and moderate brokenness of neuronal mitochondria; such mitochondrial distortion as a matter of fact appears to inspire the trademark pathologies of the illness, outstandingly amyloid-β (Aβ) plaques and hyperphosphorylated microtubule-related protein tau as neurofibrillary tangles (NFTs), and at last, appears to be liable for the trademark neurodegeneration found in AD [5]. In particular, mitochondria are related with neurodegeneration in AD for a long time, including: (1) They are the essential generators of responsive oxidative species (ROS) inside the cell [3]; (2) Damage to mitochondrial underlying parts and catalyst edifices are very much reported in AD and boundlessly go before some other trademark element of the infection [6,7,8,9]; and (3) Mitochondrial elements have been shown as seriously adjusted in AD neurons when contrasted with controls [1,5,10,11]. Since AD is the main source of decrepit dementia in the United States, influencing 15% of individuals beyond 65 years old and practically half of those more than 85 [12], the requirement for a powerful precaution measure against illness beginning and movement is truly expanding [13], and we here present current points of view on mitochondrial drugs for neurodegeneration trying to the additional helpful investigation.
Mitochondrial ROS Generation and Oxidative Stress in Alzheimer Disease: An Opportunity for Intervention
High-impact breath, representing 95% of the creature cell’s energy supply, definitely delivers receptive oxidative species inside the cell. In spite of the fact that there are systems set up to sequester such oxidants before they unleash inside the ruin, the citrus extract (TCA) cycle and the course of oxidative phosphorylation (both happening inside mitochondria) without a doubt produce an overflow of free extremists consistently. Truth be told, a few appraisals propose a day-to-day creation of 1011 ROS inside a commonplace vigorous cell [14]. As the cerebrum utilizes around 20% of the body’s oxygen supply in spite of just involving 2-3% of the weight [14], a regular grown-up neuron positively creates more. While these ROS are sensible in a youthful, solid cell, in the end, their gathering prompts cell impairments that further the phone’s failure to guard itself. Thusly, oxidative pressure turns into a critical job player in cell brokenness inside the mind, and, as a matter of fact, it has been very much recorded in neurodegenerative sicknesses, especially that of AD.
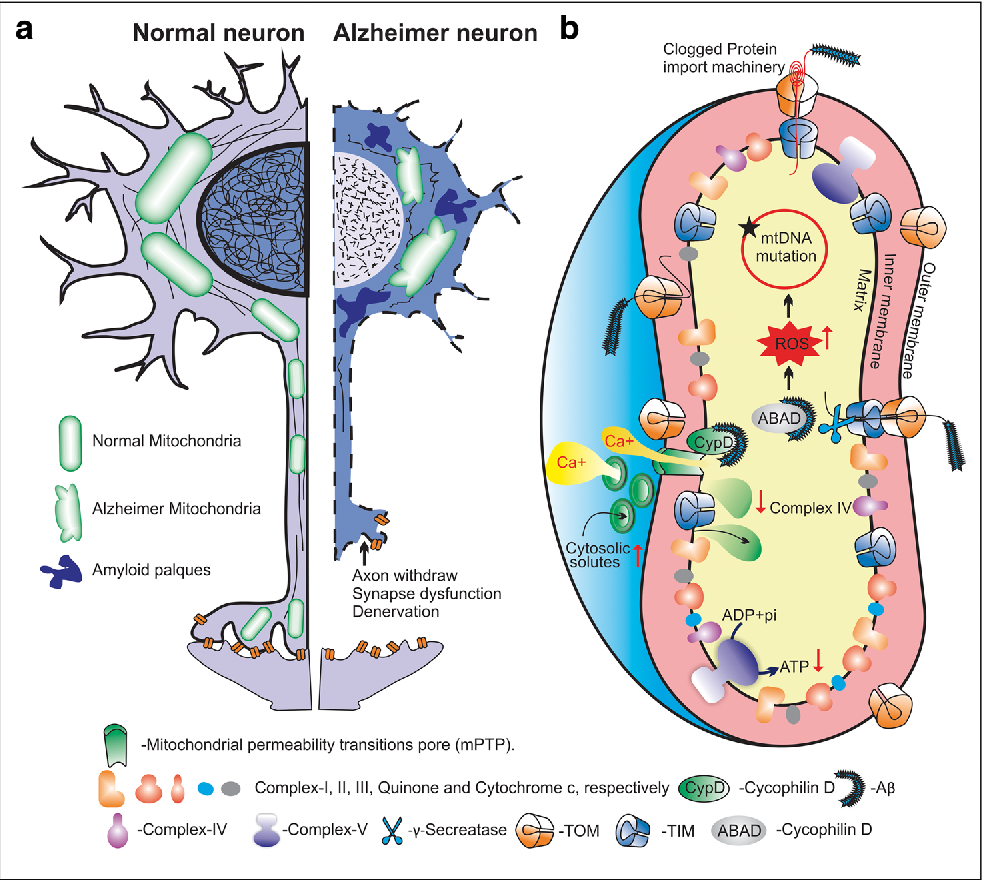
The “two-hit theory” for AD depicts a peculiarity by which the oxidative pressure inspired by broken respiratory cycles (hit one) produces compensatory changes in the cell that empower it to work for quite a long time under said pressure [8,15,16]. Sadly, these compensatory changes, including the affidavit of Aβ and phosphorylation of tau as a cancer prevention agent [17,18], make the cell helpless against extra abuses, like mitotic deviations (hit two), and it is simply the cell’s failure to shield against these second “hits” due to the compensatory “consistent express” that at last guarantees its destruction. Prominently, when oxidative harm inside the cell starts these changes (i.e., following quite a while of collection and slow oxidative harm), an endless loop results in which Aβ produces neuroinflammation and microglial initiation which, thus, evokes further oxidative pressure by means of harm to imperative mitochondrial parts, for example, pyruvate dehydrogenase complex (PDHC), ketoglutarate dehydrogenase complex (KGDH), and cytochrome c oxidase (COX) [1,19,20,21,22]. As any unsettling influence in the respiratory chemical buildings further produces ROS, in light of the fact that the mitochondrial respiratory proteins arrange the oxidation/decrease responses that empower the age of ATP, any perturbance in their instruments without a doubt prompts extra harm [23,24,25,26,27].
Extra abuses to mitochondrial parts, coming about because of oxidative pressure, have likewise been identified in AD, including modified Ca2+ homeostasis (due to mitochondrial hindrance) and mitochondrial DNA (mtDNA) transformations and cancellations [9,28,29]. As the mitochondria are the underlying maker of ROS, and in this manner of neuronal oxidative harm, and in light of the fact that mitochondria are so powerless against oxidative harms once oxidative pressure becomes wild, ongoing investigations have gone to cell reinforcement treatments in order to give a protection fix to AD [30,31,32]. Mitochondrial cancer prevention agents are of specific allure and have given the best proof of lab efficacy. Coenzyme Q10 (CoQ10) organization, for instance, has been displayed to evoke neuroprotective impacts in AD by invalidating oxidative harm and lessening mitochondrial brokenness [3,33]. Practically, CoQ10 is an electron transporter in the electron transport chain (ETC) of oxidative phosphorylation that is installed in the internal mitochondrial film. It acts to convey the high-energy electrons in the chain from complex I to complex II, and its redox cycling from the oxidized structure (ubiquinone) to the diminished structure (ubiquinol) is reliant upon ETC working [34,35]. A new report demonstrated that a portion of 6.5 μM CoQ10 to MC65 neuroblastoma cells gave total insurance from neurotoxicity because of oxidative pressure, and stifled H2O2 and O2-creation [36]. In spite of the fact that mind tissue and mitochondrial levels of CoQ10 were not expanded by the oral organization (affirmed by different reports [37,38,39], oxidative harm to cerebrum proteins was weakened [36]. Critically, the failure of orally managed cancer prevention agents to penetrate the blood cerebrum boundary (BBB) presents an obstacle in the improvement of powerful neurodegeneration therapies and has prompted the examination of more dissolvable, more limited chain cell reinforcement CoQ10 subordinates, for example, idebenone (6-(10-hydroxymethyl)- 2,3-dimethoxy-5-methyl-1,4-benzoquinone) and decylubiquinone (dUb) [40,41]. Also, mitochondria-designated cell reinforcements have been explored that further increment particularity of ROS sequestration in neurodegeneration, the best of which has been MitoQ, a triphenylphosphonium-connected ubiquinone subsidiary [42].
MitoQ is known to focus inside mitochondria (a few hundred-overlap) because of the huge mitochondrial layer potential [43]. Its particular collection in the metabolic organelle, and its consistent reusing by mitochondrial compounds (counting those of the ETC), make MitoQ a considerably more powerful cell reinforcement than those that are non-designated [44]. Without a doubt, studies play affirmed the helpful part of MitoQ in neurodegenerative models [40,45]. In particular, in parental leukemic CEM cell societies, the impacts of MitoQ were exhibited to be strikingly defensive after exhaustion of glutathione (GSH), a controller of mitochondrial porousness progress [40]. That is, MitoQ: (1) successfully hindered ROS age; (2) safeguarded mitochondrial protein redox status; (3) saved the respectability of mitochondrial constructions; and (4) impeded cell passing after exhaustion of GSH [40]. In addition, not set in stone to be a more successful cell reinforcement for mitochondria, when contrasted with CoQ10, and was exhibited to be powerful without even a trace of a working ETC [40]. While this information compares to cell societies and non-neuronal tissues, they regardless demonstrate the expected advantages of MitoQ in the treatment of oxidative pressure-related sickness. As a matter of fact, MitoQ is right now being worked on in stage II clinical preliminaries for Parkinson’s illness and liver harm related to HCV contamination [45], and the outcomes will ideally reveal insight into the pertinence of the medication to different sicknesses, like AD (Figure 1).